It is hosted both here: http://gladyshevlab.org/SelenoproteinPredictionServer/ --- by Vadim Gladyshev's lab
and here: http://seblastian.crg.es/ --- by Roderic Guigó 's lab.
SECISearch3 and Seblastian methods are described in the following paper; if the webserver is useful to your research, please cite it.
Citation:
Marco Mariotti, Alexei V. Lobanov, Roderic Guigo, and Vadim N. Gladyshev (2013) SECISearch3 and Seblastian: new tools for prediction of SECIS elements and selenoproteins.
Nucl. Acids Res. first published online June 19, 2013 doi:10.1093/nar/gkt550
Selenoproteins have been identified in all domains of life, although many lineages do not possess them and neither possess a complete Sec machinery (e.g. several insects [3]). The human selenoproteome (the set of the selenoproteins found in the genome) consists of 25 selenoproteins, mouse contains 24, fruit fly contains 3, and C.elegans just 1 [4].
For almost all known selenoproteins, a set of non-Sec containing sequence homologues are found. Almost always, these homologues contain a conserved cysteine aligned to the Sec position, and are therefore called cysteine homologues. The Sec insertion system is radically different across the 3 domains of life. The most relevant difference for this work is in the SECIS elements [5]. Eukaryotic SECIS elements are found in the 3' untranslated region (3' UTR) of selenoprotein transcripts, and possess some peculiar structural features. The structure is composed of 2 or 3 helices, with certain constraints on their length. The base of helix2 is called the SECIS core or quartet and is particularly conserved. It always includes two GA-AG pairs forming a kink turn, a peculiar RNA structural motif. At the beginning of the apical loop, the majority of eukaryotic SECIS elements possess 2 or 3 unpaired adenines, which were shown to be important for the recognition of the SECIS by the protein that binds them (SECIS binding protein 2 - SBP2).
Bacterial SECIS elements (bSECIS) are also stem loop structures, but apart from this they do not resemble their eukaryotic counterparts. Also, they are located within the coding sequence, right next to the Sec-UGA. Archael SECIS elements (aSECIS) are again stem loop structures, but different from both their bacterial and eukaryotic counterparts. Interestingly, they are located generally in the 3' UTR, with a single case found in the 5' UTR.
Given their lack of similarity, different methods must be used to search for SECIS elements in the 3 domains of life.
This web server focuses uniquely on eukaroytic selenoproteins and SECIS elements.
Due to the presence of an inframe stop codon, selenoproteins are generally misannotated in sequence databases. Thus, in the last 10 years or so some effort was spent on the development of tools dedicated to the prediction of selenoproteins. [6] [7] [8] [9] [10]
The first (original) SECISearch algorithm is described in [11] and [12], and consists of the following steps. Initially, the target sequence is scanned with Patscan, which finds matches to SECIS-like pattern. The patterns are designed to match to sequences featuring the structural elements described above. Then, RNAfold [13] is run to compute the minimal free energy structure of the sequence. Finally, a set of filters drops unstable structures and also those with features never observed in SECIS elements. Although the original SECISearch has been extremely useful for selenoprotein research, it has limitations, mainly in its dependance on the patterns and in the lack of scores assigned to output.
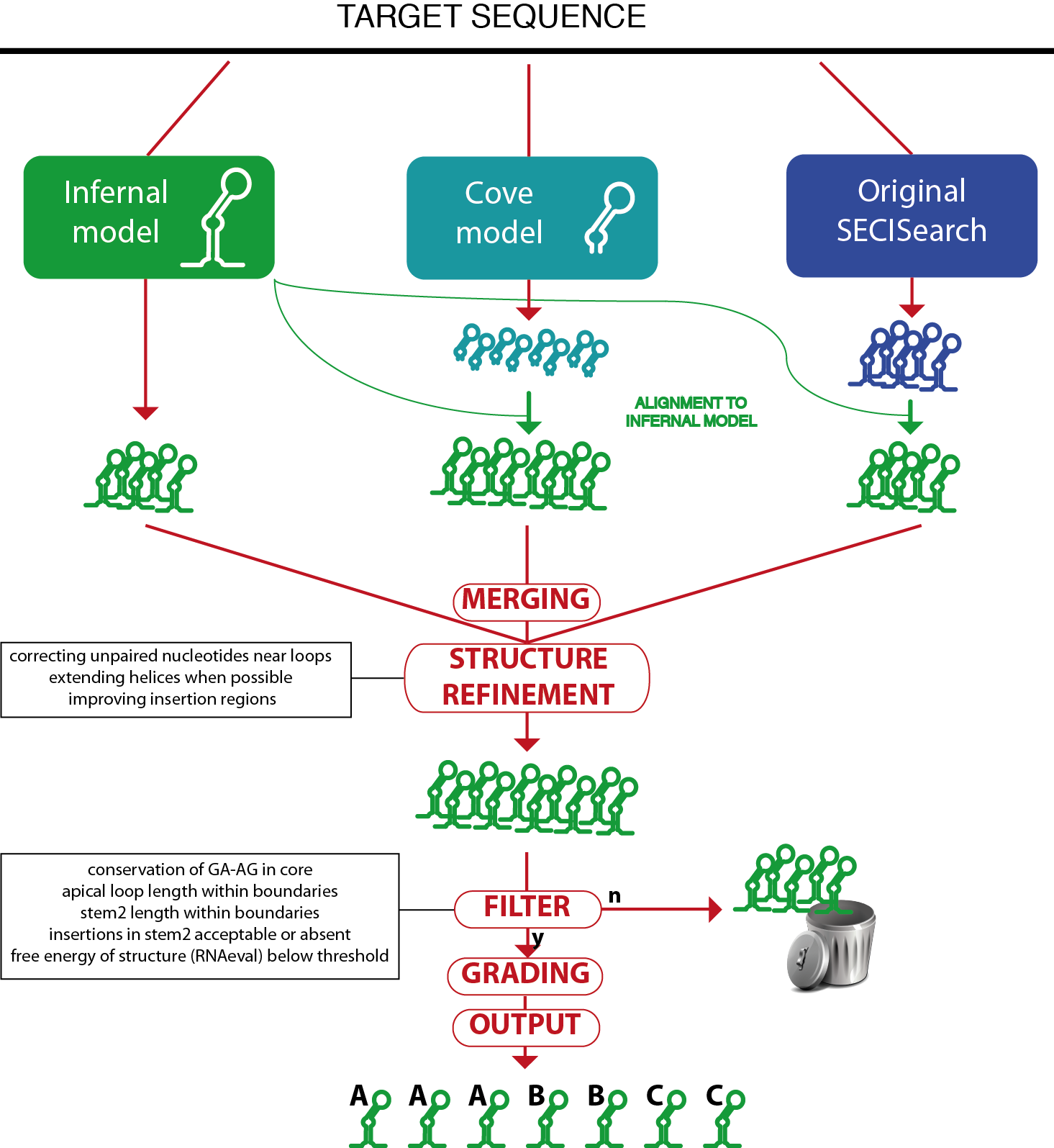
Thus, the new SECISearch3 available here has been developed. The SECISearch3 core is a model for the program Infernal [14]. The model consists of a curated, secondary structure based alignment of 1122 SECIS elements across all eukaryotic lineages. This collection of SECIS elements were predicted in the 3' UTRs of a large set of selenoproteins predicted by Selenoprofiles, a SECIS-independent pipeline for selenoprotein search.
SECISearch3 combines predictions from up to 3 sources.
The first source is Infernal: the SECIS model is used by the program cmsearch from the Infernal suite to scan the target sequence and report potential matches.
The second source is a SECIS model for the program Covels (see http://selab.janelia.org/software.html). This model was built with hundreds of manually inspected SECIS elements, but includes only the most internal part of their structure, starting from the core. The Covels matches lack a secondary structure prediction, so this is obtained through an alignment to the Infernal SECIS model using cmalign.
The third source is the original SECISearch program. To make the prediction of secondary structure uniform across the 3 methods, these predictions are also aligned to the Infernal SECIS model (the original secondary structure predicted by RNAfold is actually ignored at this point).
The detailed comparison of these 3 methods is below. Briefly, Infernal is the best method when considering both sensitivity and specificity. Covels is most sensitive method, but it is rather unspecific.
The predictions of the different sources are then merged, removing duplicates.
Then, the RNAfold package is used to compute the free energy of the structure and to refine the secondary structure prediction. Also, the Covels score is computed for all predictions, providing a way to compare SECIS predicted by different methods.
A SECIS filter is then applied to remove unprobable candidates (this can be turned off in the input page). The SECIS filter is roughly equivalent to the third stage of the original SECISearch. The filter checks the following characteristics: presence of kink-turn in the core, length of apical loop, length of stem2, bending of stem2 (insertion in 5' portion versus insertions in 3' portion), free energy of the structure.
Finally, the SECIS prediction is "graded": its sequence and structure are further analyzed by a scoring method that incoporates our experience in manual analysis of hundreds of SECIS elements. It checks several structural features of stem2 (such as the presence of consecutive mismatches, the bending of the structure, the match or mismatch state of critical positions) as well as the presence of typical unpaired nucleotides in the apical loop, as well as the Covels score for the prediction. The SECIS grade can be A, B, C in decreasing order of goodness.
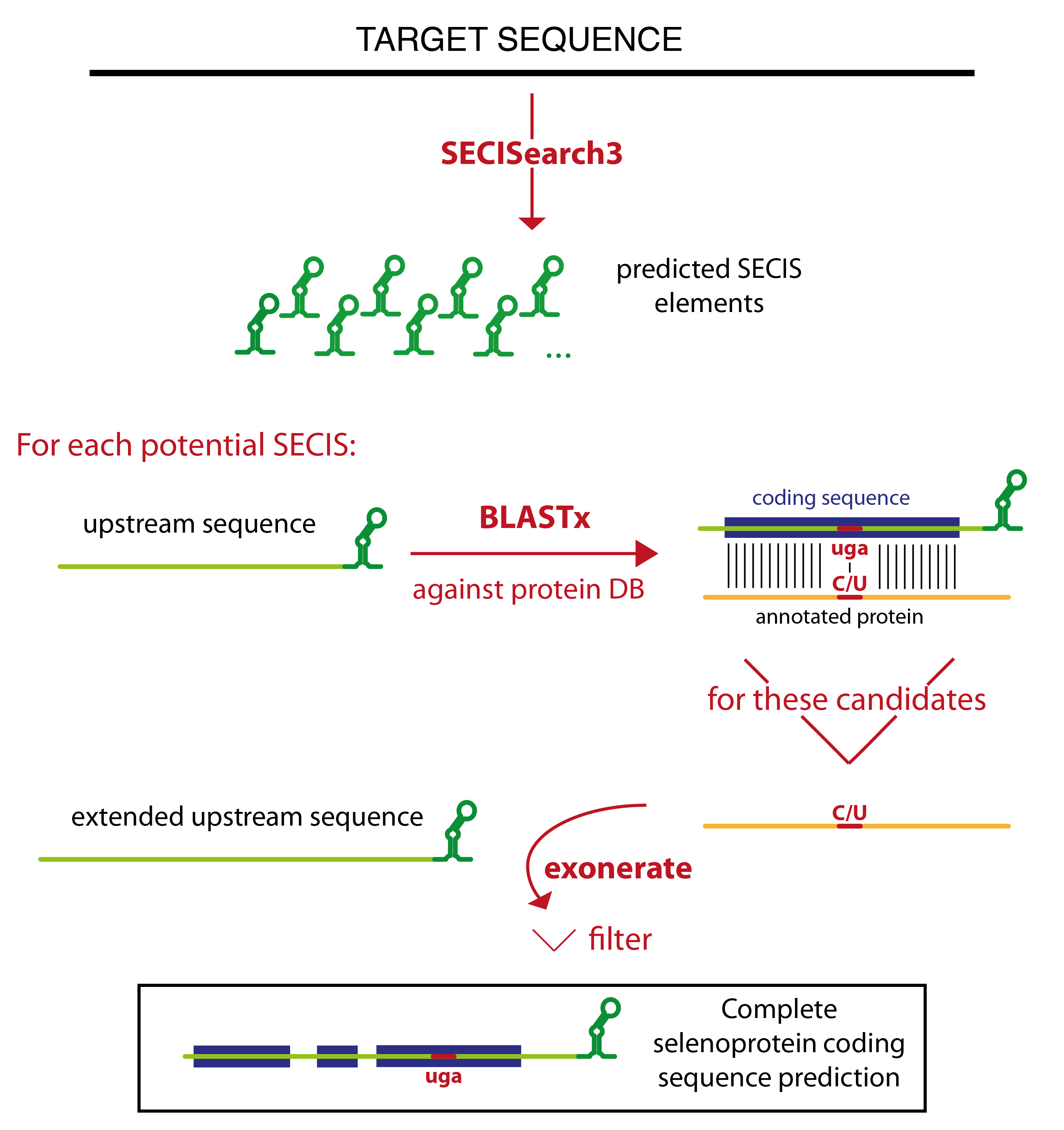
Seblastian relies on SECISearch3 for the prediction of SECIS elements. Then it extracts the sequence upstream of each potential SECIS, and runs blastx against a comprehensive protein database. The blast output is analyzed, and only certain blast hits flow through the rest of the pipeline. The best candidates are the blast hits that contain a UGA codon in the target sequence aligned to a Sec residue of an annotated selenoprotein. All other blast hits with an annotated selenoprotein (even those in which no Sec is aligned) are also considered. Then, there is a category of blast hits for the potential new selenoproteins. In these blast hits, a UGA in the target is aligned to a cysteine in an annotated protein. This strategy relies on the assumption that some cysteine homologues are already annotated. Finally, Seblastian considers also all blast hits in which the C-terminal tail of the annotated protein is aligned, since they may become candidates after the subsequent refinement of the prediction.
The program tries then to extend the gene structure of all blast hits passing this filter, joining them with other blast hits which appears to be exons of the same gene. The concept of colinearity is used: if blast hit A is found in the target downstream of blast hit B, and also the portion of the query aligned in blast hit A is downstream of that in blast hit B, they will be joined. Several blast hits may be joined in this way resulting in a multiexon gene prediction.
Exonerate is then run to refine these gene structures. The cyclic exonerate routine, described in [7], is used to ensure the identification of the whole gene structure within the exonerate detection power.
Thus, exonerate and blastx predictions are examined. Generally, exonerate prediction will be kept, but in some cases it is discarded and the gene prediction that will be output is given by blastx. This happens for example when exonerate did not output any alignment, or when the UGA previously marked as Sec candidate is not aligned.
Then, the prediction (coming from either exonerate or blast at this point) is labelled. The label identifies the type of candidate: "known selenoprotein" and "new selenoprotein" and the only ones that can be output. All predictions with a different label (for example, "no Sec candidate", or "SECIS overlapping CDS") are filtered out. In this phase, an additional filter is also applied. This filter checks that the coding sequence is not too distant from the SECIS element, but also that the candidate show a convincing patter of conservation: a number of residues conserved between query and target have to be present on both sides of the potential Sec UGA.
Seblastian can be run in two modes: search for known selenoprotein only, or search for known and new selenoproteins. In the first case, the program uses a very reduced version of the nr database containing only the annotated selenoproteins, allowing a much faster runtime.
2. Allmang C, Wurth L, Krol A(2009) The selenium to selenoprotein pathway in eukaryotes: more molecular partners than anticipated.Biochim Biophys Acta. 2009 Nov;1790(11):1415-23. Epub 2009 Mar 11. Review.
3. Chapple CE, Guigó R (2008) Relaxation of selective constraints causes independent selenoprotein extinction in insect genomes. PLoS One. 2008 Aug 13;3(8):e2968.
4. Lobanov AV, Hatfield DL, Gladyshev VN (2009) Eukaryotic selenoproteins and selenoproteomes. Biochim Biophys Acta 1790:1424-8.
5. Krol A (2002) Evolutionarily different RNA motifs and RNA-protein complexes to achieve selenoprotein synthesis. Biochimie. 2002 Aug;84(8):765-74.
6. Driscoll DM, Chavatte L (2004) Finding needles in a haystack. In silico identification of eukaryotic selenoprotein genes. EMBO Rep. 2004Feb;5(2):140-1.
7. Mariotti M, Guigó R (2010) Selenoprofiles: profile-based scanning of eukaryotic genome sequences for selenoprotein genes. Bioinformatics. 2010 Nov 1;26(21):2656-63. Epub 2010 Sep 21.
8. Jiang L, Liu Q, Ni J (2010) In silico identification of the sea squirt selenoproteome. BMC Genomics, 11, 289.
9. Li M, Huang Y, Xiao Y (2009) A method for identification of selenoprotein genes in archaeal genomes. Genomics Proteomics Bioinformatics. 2009 Jun;7(1-2):62-70.
10. Zhang,Y. and Gladyshev,V.N. (2005) An algorithm for identification of bacterial selenocysteine insertion sequence elements and selenoprotein genes. Bioinformatics, 21, 2580–2589.
11. Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigó R, Gladyshev VN (2003) Characterization of mammalian selenoproteomes. Science 300:1439-1443
12. Kryukov GV, Kryukov VM, Gladyshev VN (1999) New mammalian selenocysteine-containing proteins identified with an algorithm that searches for selenocysteine insertion sequence elements. J Biol Chem. 1999 Nov 26;274(48):33888-97
13. I.L. Hofacker, W. Fontana, P.F. Stadler, S. Bonhoeffer, M. Tacker, P. Schuster (1994) Fast Folding and Comparison of RNA Secondary Structures. Monatshefte f. Chemie 125: 167-188
14. Nawrocki EP, Kolbe DL, and Eddy SR (2009) Infernal 1.0: Inference of RNA alignments. Bioinformatics 25:1335-1337